








Transthyretin Amyloid Cardiomyopathy

Understanding the Disease Starts Here

Transthyretin Amyloid Cardiomyopathy (ATTR-CM) is a rarely diagnosed fatal heart disease affecting an estimated 400,000 people globally, although the true prevalence is unknown and may be higher.1,2 Bayer is committed to optimize the care for patients living with this progressive disease, to prolong lives, and improve patients’ quality of life.
What is Transthyretin Amyloid Cardiomyopathy (ATTR-CM)?
The disease develops when a protein that usually moves through the bloodstream and helps transport important substances becomes unstable, breaks down, and its pieces build up in the wrong organs.

Transthyretin (TTR) is a tetrameric protein formed of four subunits.3 In ATTR-CM, the tetramer destabilize into monomers.3
In patients with ATTR-CM, the protein that becomes destabilized is transthyretin (TTR). In its normal state, TTR circulates in the blood stream and plays a key role in the transport of thyroxine (a thyroid hormone) and vitamin A in the body. When it becomes destabilized, it breaks down, and its pieces form amyloid fibrils that buildup in a patient’s organs, including the heart.4
The build-up of amyloid in the heart can lead to the thickening and stiffening of the heart’s walls. This reduces the heart’s ability to effectively pump blood around the body leading to cardiomyopathy (CM), a chronic disease of the heart muscle, and eventually to heart failure.3,4
Stabilising transthyretin, and thus minimizing the formation of amyloid, is crucial to halt disease progression and improve patient outcomes.
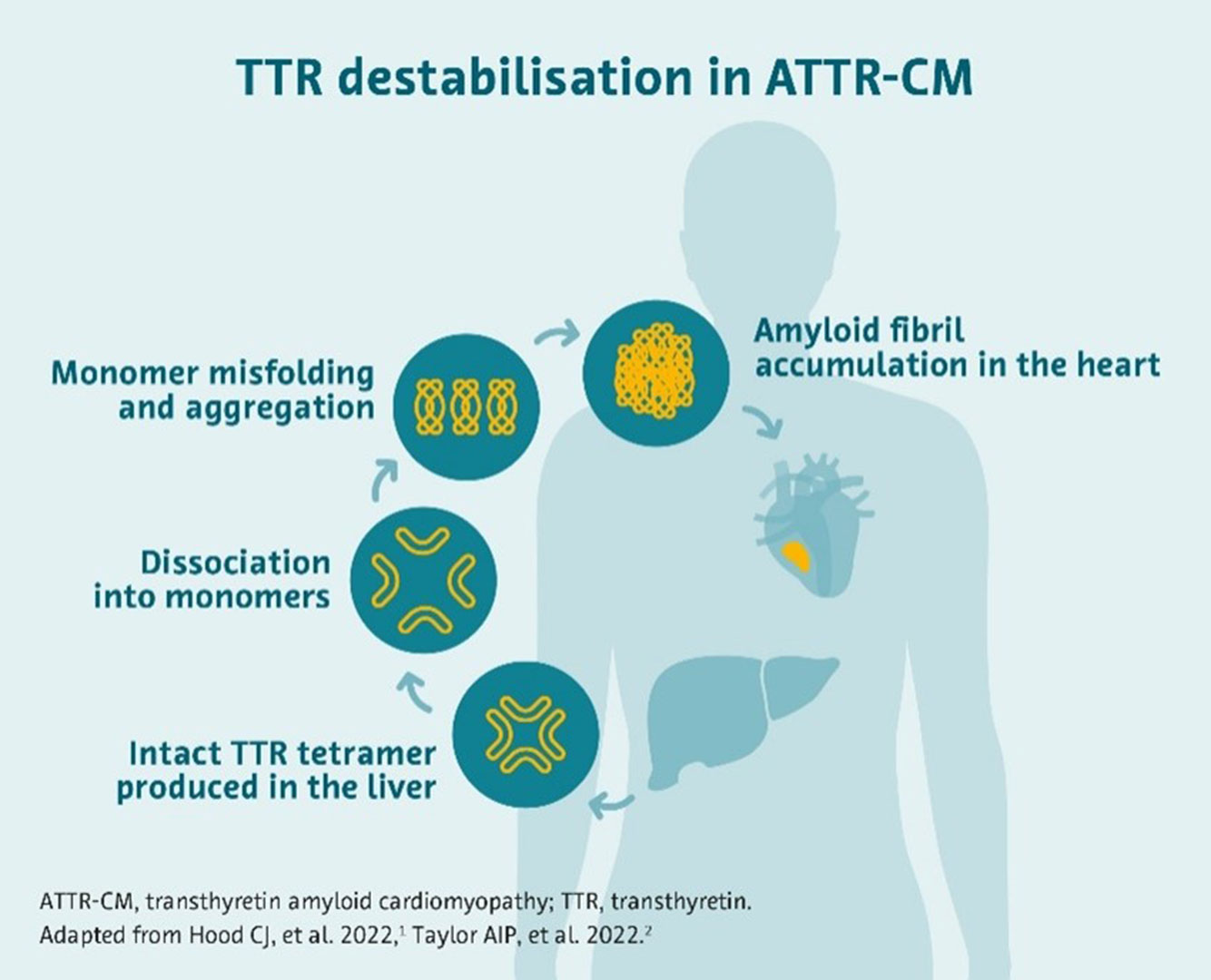
Originating from the destabilization of TTR, ATTR-CM arises from the build-up of amyloid fibrils in the heart’s walls.4,5,6
What are the signs and symptoms?
Some symptoms of ATTR-CM, including shortness of breath and chest pain, resemble those of other heart conditions,7 which can lead to delayed or misdiagnosis. Patients with ATTR-CM usually experience one or more of the following symptoms:
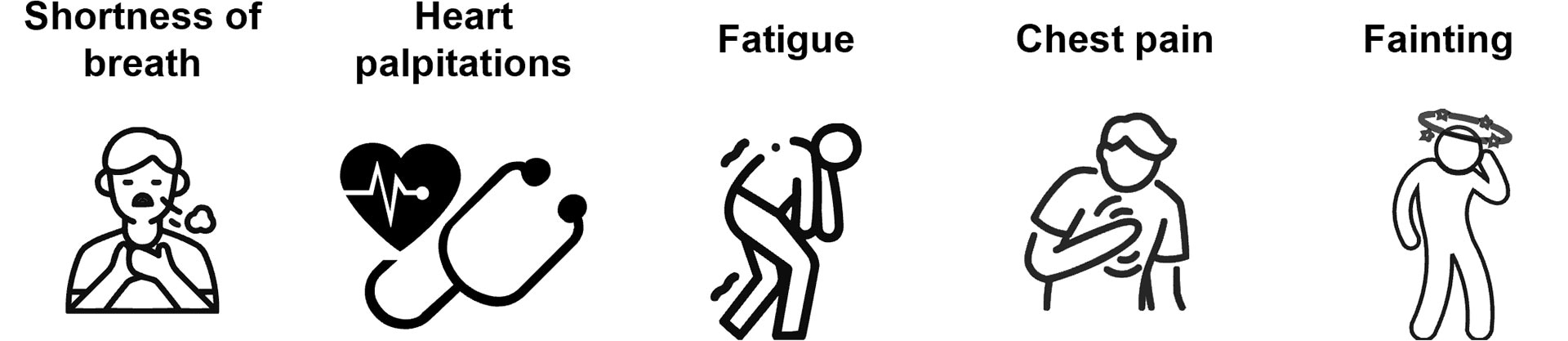
Expert consensus highlights that more effective diagnosis can be achieved through better awareness of the signs and symptoms among healthcare teams8 – improved symptom recognition in ATTR-CM may enhance patient outcomes as a result of timely diagnosis.
What are the two types of ATTR-CM?
ATTR-CM can be classified into:
- Variant ATTR-CM – also known as hereditary ATTR-CM, this is caused by an inherited mutation in the TTR gene.. It is typically seen in those aged 60 years or more, although symptoms can also develop much earlier; men are more commonly affected than women.3,9
- Wild-type ATTR-CM – this is a form of the disease which can develop as a person ages. It is most often diagnosed in men aged 65 and over.4,10
After the symptoms of ATTR-CM are identified, genetic testing is conducted to determine which gene subtype of TTR is present.11
How is ATTR-CM diagnosed?
Patients can experience years of delay before receiving a correct diagnosis when the accumulation of amyloids has already occurred, and patients are symptomatic. Notably, ATTR-CM is often misdiagnosed for hypertrophic cardiomyopathy and various other conditions due to the similarity of symptoms.13
Diagnostic delays and limited treatment options are among the main causes of poor outcomes in ATTR-CM. 14
If left untreated, these patients have a life expectancy of 2.5 to 3.5 years.15 Patients with ATTR-CM experience significantly higher mortality rates than patients with other causes of heart failure.
If patients present with symptoms common to heart failure, screening tools such as electrocardiograms, echocardiography, cardiac magnetic resonance imaging, and biomarker testing are used to determine the cause and need for further testing.16
Current and future treatment landscape
For a long time, there was no approved drug to treat ATTR-CM. Previous treatment options were limited and mainly focus on symptom management and supportive care.7
In recent years, diagnosis and treatment management has improved, and the disease is diagnosed and managed at earlier disease stages, and sometimes before severe progression.12,17
But there is still high unmet need in terms of survival, hospitalisation and quality of life.

Our commitment in the field of ATTR-CM
At Bayer, it is our mission to address patients’ needs and to push the boundaries of innovation.
We are dedicated to improving patient outcomes in heart disease. Our broad heart disease portfolio represents strategic investments aimed at addressing the substantial morbidity, mortality, and economic burden associated with heart disease.
Building on our strong legacy in cardiology, we are committed to improving the care for patients suffering from ATTR-CM and advancing their treatment options.
1Kim M, et al. Comparative Outcomes of a Transthyretin Amyloid Cardiomyopathy Cohort Versus Patients With Heart Failure With Preserved Ejection Fraction Enrolled in the TOPCAT Trial. Journal of the American Heart Association. 2023;12(15). https://doi.org/10.1161/JAHA.123.029705.
2ClinicalTrials.gov. Efficacy and Safety of AG10 in Subjects With Transthyretin Amyloid Cardiomyopathy (ATTRibute-CM). Available at: https://clinicaltrials.gov/study/NCT03860935. [Accessed 01 October 2024]
3Ruberg FL, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. Journal of the American College of Cardiology. 2019;73(22):2872-91.
4Kittleson MM, et al. American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7-22.
5Hood CJ, et al. Update on disease-specific biomarkers in transthyretin cardiac amyloidosis. Current heart failure reports. 2022 Oct;19(5):356-63.
6Taylor AI, Staniforth RA. General principles underpinning amyloid structure. Frontiers in Neuroscience. 2022 Jun 2;16:878869.
7NHS. First ever life-saving treatment for rare heart condition available on the NHS. Available at: https://www.england.nhs.uk/2024/05/first-ever-life-saving-treatment-for-rare-heart-condition-available-on-the-nhs/. [Accessed 01 October 2024]
8Garcia‐Pavia P, et al. Expert consensus on the monitoring of transthyretin amyloid cardiomyopathy. European journal of heart failure. 2021;23(6):895-905.
9Gertz M. Hereditary ATTR Amyloidosis: Burden of Illness and Diagnostic Challenges. American Journal of Managed Care. 2017;23(7 Suppl):S107-S112
10Jain A, Zahra F. Transthyretin amyloid cardiomyopathy (ATTR-CM). StatPearls Publishing. 2023.
11Benson MD, et al. Diagnosis and screening of patients with hereditary transthyretin amyloidosis (hATTR): current strategies and guidelines. Therapeutics and Clinical Risk Management. 2020:749-58.
12Lane T, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16-26.
13Gertz M, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC family practice. 2020;21:1-2.
14Tschöpe C, Elsanhoury A. Treatment of transthyretin amyloid cardiomyopathy: the current options, the future, and the challenges. Journal of Clinical Medicine. 2022;11(8):2148.
15Gang S, et al. Clinical manifestation, economic burden, and mortality in patients with transthyretin cardiac amyloidosis. Orphanet Journal of Rare Diseases. 2022;17(1).
16Maurer MS, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circulation: Heart Failure. 2019;12(9):e006075.
17Ioannou A, et al. Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation. 2022 Nov 29;146(22):1657-70.